

SARS-CoV-2 è il settimo coronavirus noto per infettare l’uomo; SARS-CoV, MERS-CoV e SARS-CoV-2 possono causare gravi malattie, mentre HKU1, NL63, OC43 e 229E sono associati a sintomi lievi. Qui passiamo in rassegna cosa si può dedurre sull’origine di SARS-CoV-2 dall’analisi comparativa dei dati genomici. Offriamo una prospettiva sulle notevoli caratteristiche del genoma SARS-CoV-2 e discutiamo gli scenari in base ai quali potrebbero essere sorti. Le nostre analisi mostrano chiaramente che SARS-CoV-2 non è un costrutto di laboratorio o un virus appositamente manipolato.
Caratteristiche notevoli del genoma SARS-CoV-2
1. Mutazioni nel dominio del recettore di SARS-CoV-2
Il dominio legante il recettore (RBD) nella proteina di picco è la parte più variabile del genoma del coronavirus. Sei aminoacidi RBD hanno dimostrato di essere critici per il legame con i recettori ACE2 e per determinare l’intervallo ospite di virus simili a SARS-CoV 7. Con coordinate basate su SARS-CoV, sono Y442, L472, N479, D480, T487 e Y4911, che corrispondono a L455, F486, Q493, S494, N501 e Y505 in SARS-CoV-2. Cinque di questi sei residui differiscono tra SARS-CoV-2 e SARS-CoV (Fig. 1a ). Sulla base di studi strutturali ed esperimenti biochimici, SARS-CoV-2 sembra avere un RBD che si lega con elevata affinità con ACE2 di esseri umani, furetti, gatti e altre specie con elevata omologia dei recettori.
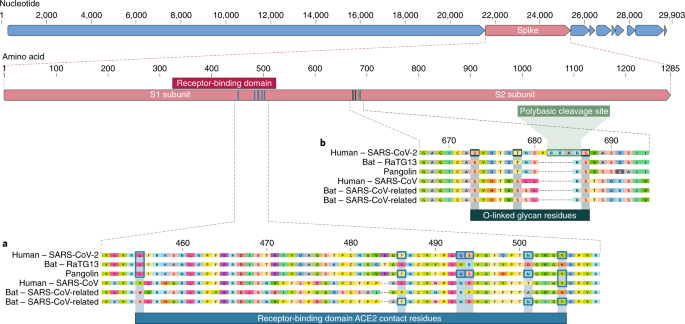
Mentre le analisi sopra suggeriscono che SARS-CoV-2 può legare ACE2 umano con elevata affinità, le analisi computazionali prevedono che l’interazione non è l’ideale e che la sequenza RBD è diversa da quelle mostrate in SARS-CoV per essere ottimale per il legame del recettore. Pertanto, il legame ad alta affinità della proteina di picco SARS-CoV-2 con l’ACE2 umano è molto probabilmente il risultato della selezione naturale su un ACE2 umano o simile all’uomo che consente la formazione di un’altra soluzione di legame ottimale. Questa è una prova evidente che SARS-CoV-2 non è il prodotto di una manipolazione intenzionale.
2. Sito di scissione della furina polibasica e glicani O-linked
La seconda caratteristica notevole di SARS-CoV-2 è un sito di scissione polibasico (RRAR) alla giunzione di S1 e S2, le due subunità del picco (Fig. 1b ). Ciò consente una scissione efficace da parte della furina e di altre proteasi e ha un ruolo nel determinare l’infettività virale e l’intervallo dell’ospite. Inoltre, in questo sito è inserito anche un prolina principale in SARS-CoV-2; quindi, la sequenza inserita è PRRA (Fig. 1b ). Si prevede che la svolta creata dalla prolina porti all’aggiunta di glicani O-linked a S673, T678 e S686, che fiancheggiano il sito di scissione e sono unici per SARS-CoV-2 (Fig. 1b). Non sono stati osservati siti di scissione polibasici nei relativi betacoronavirus “lineage B”, sebbene altri betacoronavirus umani, incluso HKU1 (lineage A), abbiano tali siti e predissero glicani O-linked. Dato il livello di variazione genetica nel picco, è probabile che virus simili a SARS-CoV-2 con siti di scissione polibasici parziali o completi saranno scoperti in altre specie.
La conseguenza funzionale del sito di scissione polibasico in SARS-CoV-2 non è nota e sarà importante determinarne l’impatto sulla trasmissibilità e sulla patogenesi nei modelli animali. Esperimenti con SARS-CoV hanno dimostrato che l’inserimento di un sito di scissione della furina nella giunzione S1 – S2 migliora la fusione cellula-cellula senza influenzare l’ingresso virale. Inoltre, la scissione efficiente del picco MERS-CoV consente ai coronavirus MERS-like dei pipistrelli di infettare le cellule umane. Nei virus dell’influenza aviaria, la replicazione e la trasmissione rapide in popolazioni di polli ad alta densità selezionano per l’acquisizione di siti di scissione polbasici nella proteina di emoagglutinina (HA), che svolge una funzione simile a quella della proteina spike dei coronavirus. L’acquisizione di siti polivinici di scissione in HA, mediante inserimento o ricombinazione, converte i virus dell’influenza aviaria a bassa patogenicità in forme altamente patogene. L’acquisizione di siti di scissione polibasici da parte di HA è stata osservata anche dopo ripetuti passaggi nella coltura cellulare o attraverso animali.
La funzione dei glicani O-predetti previsti non è chiara, ma potrebbero creare un “dominio simile alla mucina” che protegge gli epitopi o i residui chiave sulla proteina del picco SARS-CoV-2. Numerosi virus utilizzano domini simili a quelli della mucina in quanto gli scudi di glicano coinvolgono l’immunoevasione. Sebbene la previsione della glicosilazione legata all’O sia solida, sono necessari studi sperimentali per determinare se questi siti sono utilizzati in SARS-CoV-2.
Teorie delle origini SARS-CoV-2
1. Selezione naturale in un ospite animale prima del trasferimento zoonotico
Poiché molti dei primi casi di COVID-19 erano collegati al mercato huanan di Wuhan, è possibile che in questa località fosse presente una fonte animale. Data la somiglianza di SARS-CoV-2 con i coronavirus simili a SARS-CoV, è probabile che i pipistrelli fungano da ospiti di riserva per il suo progenitore. Sebbene RaTG13, prelevato da un pipistrello Rhinolophus affinis, sia ~ 96% complessivamente identico a SARS-CoV-2, il suo picco diverge nell’RBD, il che suggerisce che potrebbe non legarsi efficacemente all’ACE2 umano (Fig. 1a ).
I pangolini malesi (Manis javanica ) importati illegalmente nella provincia del Guangdong contengono coronavirus simili a SARS-CoV-2. Sebbene il virus del pipistrello RaTG13 rimanga il più vicino al SARS-CoV-2 nel genoma, alcuni coronavirus del pangolino mostrano una forte somiglianza con il SARS-CoV-2 nell’RBD, inclusi tutti e sei i residui chiave dell’RBD (Fig. 1 ). Ciò dimostra chiaramente che la proteina di picco SARS-CoV-2 ottimizzata per il legame con ACE2 simile all’uomo è il risultato della selezione naturale.
Né i betacoronavirus di pipistrello né i betacoronavirus di pangolino campionati finora hanno siti di scissione polibasici. Sebbene non sia stato identificato alcun coronavirus animale sufficientemente simile da essere il progenitore diretto della SARS-CoV-2, la diversità dei coronavirus nei pipistrelli e in altre specie è fortemente sottocampionata. Mutazioni, inserzioni ed eliminazioni possono verificarsi vicino alla giunzione S1-S2 dei coronavirus, che mostra che il sito di scissione polibasico può derivare da un processo evolutivo naturale. Affinché un virus precursore acquisisca sia il sito di scissione polibasico sia le mutazioni nella proteina spike adatte per legarsi all’ACE2 umano, un ospite animale dovrebbe probabilmente avere un’alta densità di popolazione (per consentire alla selezione naturale di procedere in modo efficiente) e una codifica ACE2 gene simile all’ortoologo umano.
2. Selezione naturale nell’uomo a seguito di trasferimento zoonotico
È possibile che un progenitore della SARS-CoV-2 sia saltato nell’uomo, acquisendo le caratteristiche genomiche sopra descritte attraverso l’adattamento durante la trasmissione da uomo a uomo non rilevata. Una volta acquisiti, questi adattamenti consentirebbero alla pandemia di decollare e produrre un gruppo di casi sufficientemente ampio da innescare il sistema di sorveglianza che lo ha rilevato.
Tutti i genomi SARS-CoV-2 sequenziati finora hanno le caratteristiche genomiche sopra descritte e sono quindi derivati da un antenato comune che li aveva. La presenza nei pangolini di un RBD molto simile a quello della SARS-CoV-2 significa che possiamo dedurre che così era probabilmente anche nel virus che è saltato nell’uomo. Questo lascia l’inserimento del sito di scissione polibasico durante la trasmissione da uomo a uomo.
Le stime dei tempi del più recente antenato comune di SARS-CoV-2 effettuate con i dati attuali della sequenza indicano l’emergenza del virus tra la fine di novembre 2019 e l’inizio di dicembre 2019, compatibile con i primi casi confermati retrospettivamente. Quindi, questo scenario presume un periodo di trasmissione non riconosciuta nell’uomo tra l’evento zoonotico iniziale e l’acquisizione del sito di scissione polibasico. Occorrerebbero opportunità sufficienti se ci fossero stati molti eventi zoonotici precedenti che hanno prodotto brevi catene di trasmissione da uomo a uomo per un lungo periodo. Questa è essenzialmente la situazione per MERS-CoV, per la quale tutti i casi umani sono il risultato di ripetuti salti del virus da cammelli dromedari, producendo singole infezioni o brevi catene di trasmissione che alla fine si risolvono, senza adattamento alla trasmissione sostenuta.
Gli studi sui campioni umani bancari potrebbero fornire informazioni sull’eventuale diffusione criptica. Anche studi sierologici retrospettivi potrebbero essere istruttivi e alcuni di questi studi sono stati condotti mostrando esposizioni a basso livello a coronavirus SARS-CoV-like in alcune aree della Cina. Criticamente, tuttavia, questi studi non avrebbero potuto distinguere se le esposizioni fossero dovute a infezioni precedenti con SARS-CoV, SARS-CoV-2 o altri coronavirus simili a SARS-CoV. Ulteriori studi sierologici devono essere condotti per determinare l’estensione della precedente esposizione umana alla SARS-CoV-2.
3. Selezione durante il passaggio
La ricerca di base che coinvolge il passaggio di coronavirus di tipo SARS-CoV di tipo pipistrello in colture cellulari e / o modelli animali è in corso da molti anni in laboratori di livello 2 di biosicurezza in tutto il mondo e ci sono casi documentati di fughe di laboratorio di SARS-CoV. Dobbiamo quindi considerare la possibilità di un rilascio involontario di laboratorio di SARS-CoV-2.
In teoria, è possibile che SARS-CoV-2 abbia acquisito mutazioni RBD (Fig. 1a ) durante l’adattamento al passaggio nella coltura cellulare, come è stato osservato negli studi di SARS-CoV.
La scoperta di coronavirus SARS-CoV-like da pangolini con RBD quasi identici, tuttavia, fornisce una spiegazione molto più forte e più parsimoniosa di come SARS-CoV-2 li ha acquisiti tramite ricombinazione o mutazione.
L’acquisizione sia del sito di clivaggio polibasico sia dei glicani O-linked previsti contesta anche scenari basati sulla cultura. Nuovi siti di scissione poliassici sono stati osservati solo dopo un passaggio prolungato del virus dell’influenza aviaria a bassa patogenicità in vitro o in vivo. Inoltre, un’ipotetica generazione di SARS-CoV-2 per coltura cellulare o passaggio di animali avrebbe richiesto l’isolamento preventivo di un virus progenitore con somiglianza genetica molto elevata, che non è stata descritta. La successiva generazione di un sito di scissione polibasica avrebbe quindi richiesto un passaggio ripetuto nella coltura cellulare o in animali con recettori ACE2 simili a quelli umani, ma tale lavoro non è stato precedentemente descritto. Infine, è improbabile che la generazione dei glicani O-predetti si sia verificata a causa del passaggio della coltura cellulare, poiché tali caratteristiche suggeriscono il coinvolgimento di un sistema immunitario.
conclusioni
Le caratteristiche genomiche descritte qui possono spiegare in parte l’infettività e la trasmissibilità della SARS-CoV-2 nell’uomo. Sebbene le prove dimostrino che SARS-CoV-2 non è un virus manipolato intenzionalmente, attualmente è impossibile provare o confutare le altre teorie sulla sua origine descritte qui. Tuttavia, poiché abbiamo osservato tutte le notevoli caratteristiche di SARS-CoV-2, incluso l’RBD ottimizzato e il sito di scissione polibasico, in coronavirus correlati in natura, non crediamo che qualsiasi tipo di scenario di laboratorio sia plausibile.
Ulteriori dati scientifici potrebbero far oscillare l’equilibrio delle prove per favorire un’ipotesi rispetto a un’altra. Ottenere sequenze virali correlate da fonti animali sarebbe il modo più definitivo per rivelare le origini virali. Ad esempio, una futura osservazione di un sito di scissione polibasico intermedio o completamente formato in un virus simile a SARS-CoV-2 negli animali darebbe un ulteriore supporto alle ipotesi di selezione naturale. Sarebbe anche utile ottenere ulteriori dati genetici e funzionali sulla SARS-CoV-2, inclusi studi sugli animali. L’identificazione di un potenziale ospite intermedio di SARS-CoV-2, così come il sequenziamento del virus da casi molto precoci, sarebbe allo stesso modo altamente informativo. Indipendentemente dagli esatti meccanismi con cui SARS-CoV-2 ha avuto origine mediante selezione naturale.





































